Openness and collaboration go hand in hand. Scientists at PNNL are working with the Frictionless Data team at Open Knowledge International to ensure collaboration on data analysis is seamless and their data integrity is maintained.
I’m a computational biologist at the Pacific Northwest National Laboratory (PNNL), where I work on environmental and biomedical research. In our scientific endeavors, the full data life cycle typically involves new algorithms, data analysis and data management. One of the unique aspects of PNNL as a U.S. Department of Energy National Laboratory is that part of our mission is to be a resource to the scientific community. In this highly collaborative atmosphere, we are continuously engaging research partners around the country and around the world.
 Image credit: unsplash (public domain)
Image credit: unsplash (public domain)
One of my recent research topics is how to make collaborative data analysis more efficient and more impactful. In most of my collaborations, I work with other scientists to analyze their data and look for evidence that supports or rejects a hypothesis. Because of my background in computer science, I saw many similarities between collaborative data analysis and collaborative software engineering. This led me to wonder, “We use version control for all our software products. Why don’t we use version control for data analysis?” This thought inspired my current project and has prompted other open data advocates like Open Knowledge International to propose source control for data.
Openness is a foundational principle of collaboration. To work effectively as a team, people need to be able to easily see and replicate each other’s work. In software engineering, this is facilitated by version control systems like Git or SVN. Version control has been around for decades and almost all best practices for collaborative software engineering explicitly require version control for complete sharing of source code within the development team. At the moment we don’t have a similarly ubiquitous framework for full sharing in data analysis or scientific investigation. To help create this resource, we started Active Data Biology. Although the tool is still in beta-release, it lays the groundwork for open collaboration.
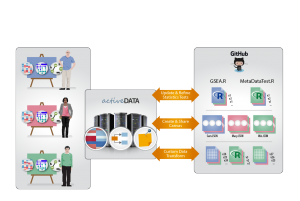
The original use case for Active Data Biology is to facilitate data analysis of gene expression measurements of biological samples. For example, we use the tool to investigate the changing interaction of a bacterial community over time; another great example is the analysis of global protein abundance in a collection of ovarian tumors. In both of these experiments, the fundamental data consist of two tables: 1) a matrix of gene expression values for each sample; 2) a table of metadata describing each sample. Although the original instrument files used to generate these two simple tables are often hundreds of gigabytes, the actual tables are relatively small.
To work effectively as a team, people need to be able to easily see and replicate each other’s work.
After generating data, the real goal of the experiment is to discover something profoundly new and useful – for example how bacteria growth changes over time or what proteins are correlated with surviving cancer. Such broad questions typically involve a diverse team of scientists and a lengthy and rigorous investigation. Active Data Biology uses version control as an underlying technology to ease collaboration between these large and diverse groups.
 Active Data Biology creates a repository for each data analysis project. Inside the repository live the data, analysis software, and derived insight. Just as in software engineering, the repository is shared by various team members and analyses are versioned and tracked over time. Although the framework we describe here was created for our specific biological data application, it is possible to generalize the idea and adapt it to many different domains.
Active Data Biology creates a repository for each data analysis project. Inside the repository live the data, analysis software, and derived insight. Just as in software engineering, the repository is shared by various team members and analyses are versioned and tracked over time. Although the framework we describe here was created for our specific biological data application, it is possible to generalize the idea and adapt it to many different domains.
An example repository can be found here. This dataset originates from a proteomics study of ovarian cancer. In total, 174 tumors were analyzed to identify the abundance of several thousand proteins. The protein abundance data is located in this repository. In order to more easily analyze this with our R based statistical code, we also store the data in an Rdata file (data.Rdata). Associated with this data file is a metadata table which describes the tumor samples, e.g. age of the patient, tumor stage, chemotherapy status, etc. It can be found at metadata.tsv (For full disclosure, and to calm any worries, all of the samples have been de-identified and the data is approved for public release.)
Data analysis is an exploration of data, an attempt to uncover some nugget which confirms a hypothesis. Data analysis can take many forms. For me it often involves statistical tests which calculate the likelihood of an observation. For example, we observe that a set of genes which have a correlated expression pattern and are enriched in a biological process. What is the chance that this observation is random? To answer this, we use a statistical test (e.g. a Fisher’s exact test). As the specific implementation might vary from person to person, having access to the exact code is essential. There is no “half-way” sharing here. It does no good to describe analyses over the phone or through email; your collaborators need your actual data and code.
In Active Data Biology, analysis scripts are kept in the repository. This repository had a fairly simple scope for statistical analysis. The various code snippets handled data ingress, dealt with missing data (a very common occurrence in environmental or biomedical data), performed a standard test and returned the result. Over time, these scripts may evolve and change. This is exactly why we chose to use version control, to effortlessly track and share progress on the project.
We should note that we are not the only ones using version control in this manner. Open Knowledge International has a large number of GitHub repositories hosting public datasets, such as atmospheric carbon dioxide time series measurements. Vanessa Bailey and Ben Bond-Lamberty, environmental scientists at PNNL, used GitHub for an open experiment to store data, R code, a manuscript and various other aspects of analysis. The FiveThirtyEight group, led by Nate Silver, uses GitHub to share the data and code behind their stories and statistical exposés. We believe that sharing analysis in this way is critical for both helping your team work together productively and also for gaining public trust.
At PNNL, we typically work in a team that includes both computational and non-computational scientists, so we wanted to create an environment where data exploration does not necessarily require computational expertise. To achieve this, we created a web-based visual analytic which exposes the data and capabilities within a project’s GitHub repository. This gives non-computational researchers a more accessible interface to the data, while allowing them access to the full range of computational methods contributed by their teammates. We first presented the Active Data Biology tool at Nature’s Publishing Better Science through Better Data conference. It was here that we met Open Knowledge International. Our shared passion for open and collaborative data through tools like Git led to a natural collaboration. We’re excited to be working with them on improving access to scientific data and results.
![]() On the horizon, we are working together to integrate Frictionless Data and Good Tables into our tool to help validate and smooth our data access. One of the key aspects of data analysis is that it is fluid; over the course of investigation your methods and/or data will change. For that reason, it is important that the data integrity is always maintained. Good Tables is designed to enforce data quality; consistently verifying the accuracy of our data is essential in a project where many people can update the data.
On the horizon, we are working together to integrate Frictionless Data and Good Tables into our tool to help validate and smooth our data access. One of the key aspects of data analysis is that it is fluid; over the course of investigation your methods and/or data will change. For that reason, it is important that the data integrity is always maintained. Good Tables is designed to enforce data quality; consistently verifying the accuracy of our data is essential in a project where many people can update the data.
One of the key aspects of data analysis is that it is fluid…For that reason, it is important that the data integrity is always maintained.
One of our real-world problems is that clinical data for biomedical projects is updated periodically as researchers re-examine patient records. Thus the meta-data describing a patient’s survival status or current treatments will change. A second challenge discovered through experience is that there are a fair number of entry mistakes, typos or incorrect data formatting. Working with the Open Knowledge International team, we hope to reduce these errors at their origin by enforcing data standards on entry, and continuously throughout the project.
I look forward to data analysis having the same culture as software engineering, where openness and sharing has become the norm. To get there will take a bit of education as well as working out some standard structures/platforms to achieve our desired goal.
Dr. Payne's research interests are focused on algorithms for proteomics data analysis and subsequent interpretation and integration.
He is a DOE Early Career Investigator for algorithmic research in metaproteomics, or proteomics of environmental communities. Additional projects include data visualization and proteome/genome integration.
Prior to joining PNNL, Dr. Payne was an Assistant Professor of Informatics at the J. Craig Venter Institute. Dr. Payne received a B.S. of computer science at Brigham Young University. He earned his Ph.D. in Bioinformatics from UC, San Diego.







